Wilson disease is a genetic disorder that is fatal unless detected and treated before serious illness from copper poisoning develops. Wilson disease affects approximately one in 30,000 people worldwide. The genetic defect causes excessive copper accumulation in the liver or brain.
Small amounts of the mineral copper are as essential as vitamins in a healthy body. Copper is present in many foods ranging from shellfish to vegetables, so most people consume more dietary copper than they need. Healthy people excrete the copper they do not need, but Wilson disease patients cannot.
Copper begins to accumulate immediately after birth. Over time, the excess copper poisons the liver or brain, resulting in liver, neurologic or psychiatric symptoms. The symptoms usually appear in late adolescence to early adulthood but can occur in early childhood and sometimes even in middle or old age.
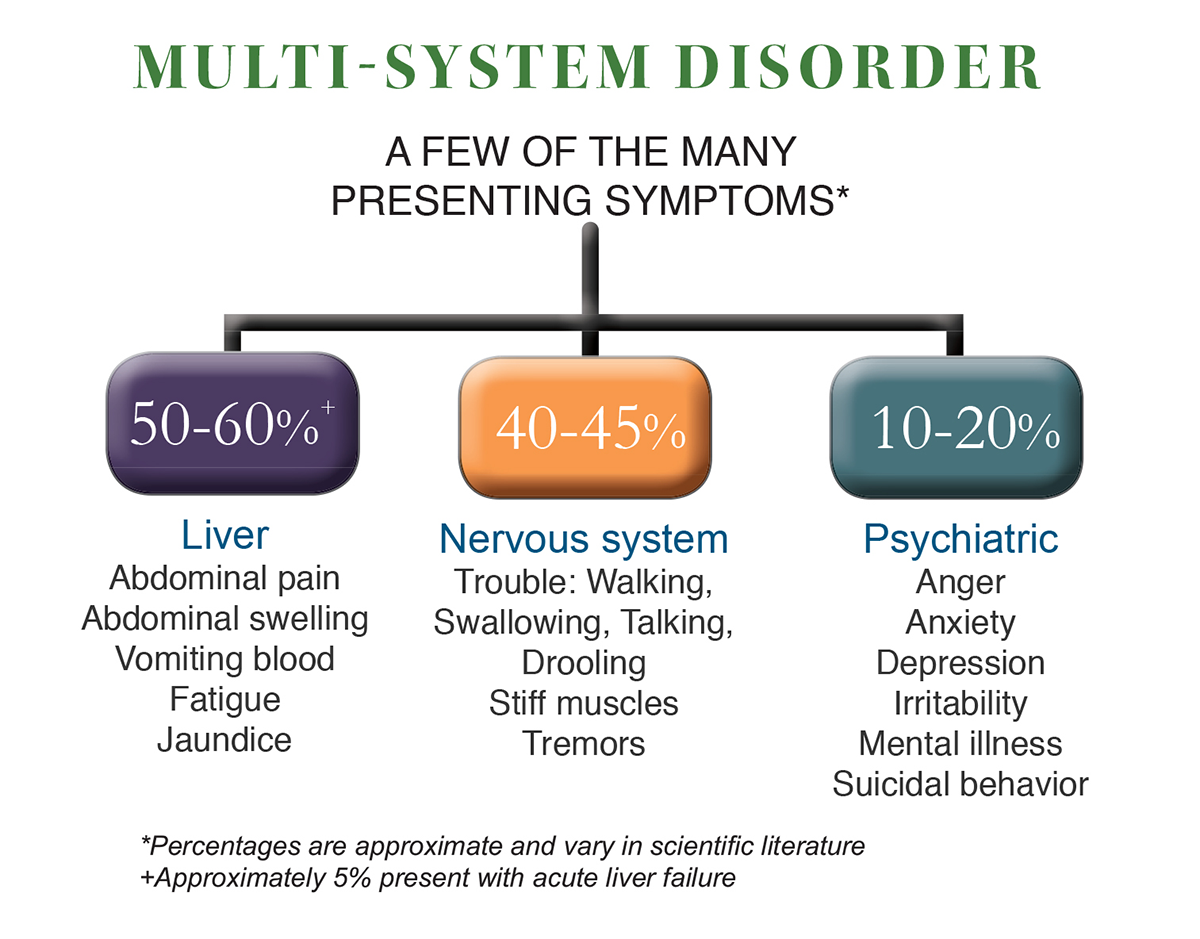
The first signs that the liver is being attacked may be yellowing of the skin or eyes, abdominal swelling, vomiting of blood, and abdominal pain. When copper attacks the brain, the early neurological signs may include changes in handwriting, drooling, tremors, and difficulty walking, talking, and swallowing. Some people have psychological or behavioral changes that may include depression, anxiety, impulsivity, and aggression. Women may have menstrual irregularities, absence of periods, infertility, or multiple miscarriages. No matter how the disease begins, it is always fatal if it is not diagnosed and treated with a daily medication regimen for life.
The first part of the body that copper affects is the liver. In about half of Wilson disease patients the liver is the only affected organ. The initial physical changes in the liver can be detected through a liver biopsy or a fibroscan test. When signs of liver disease develop, patients are often thought to have infectious hepatitis or infectious mononucleosis when they actually have Wilson disease. Testing for Wilson disease should be performed in individuals with unexplained, abnormal liver tests.
Some WD patients do not show any signs of liver disease. Neurological changes are the first sign In about forty percent of patients. When neurological symptoms cannot be explained by other disorders such as Parkinson’s disease, then Wilson disease should be considered. If unexplained symptoms are present and do not get better with typical therapies, and there is evidence of liver disease in lab testing, then a WD diagnosis must be considered.
Up to twenty percent of patients will have seen a psychiatrist before getting a diagnosis of Wilson disease. Most often, these patients will have mood issues such as depression and bipolar disorders, including anxiety, Obsessive-Compulsive Disorder (OCD), and Attention Deficit Hyperactivity Disorder (ADHD). Young patients may be misdiagnosed as poor school performers.
A simple eye exam with a slit-lamp can pick up a sign of Wilson disease called Kayser-Fleischer (KF) rings. These copper deposits in the cornea can be seen in about 95% of patients with neurological and/or psychiatric symptoms, but only in about half of those with signs of liver disease.